Introduction
Dear nf-core community, the core team would like to inform you about some upcoming changes in how we would like to handle software containers… 📦
Software containers are a fundamental part of modern bioinformatics workflows. Nextflow supports multiple container platforms, but the two most commonly used are Docker and Singularity. When using containers, all software requirements for a specific Nextflow process are wrapped up into an image and referenced within the pipeline code. The process tasks run in isolation in the host environment, and the end user doesn’t need to worry about installing software dependencies for the pipeline. The software builds are locked in time, making results highly reproducible over many years.
To ensure maximum compatibility across different environments, nf-core pipelines ship with the full range of options: conda environments as well as Docker and Singularity images. These are typically built using Bioconda and BioContainers. The Bioconda and BioContainer projects have been invaluable to nf-core’s success. We’re hugely grateful to all contributors for their work, as well as to Anaconda, Quay.io and the Galaxy project for hosting these resources.
Change is on the breeze

Bioconda and BioContainers have worked very well for nf-core. However, we are now looking to migrate to a new system: Seqera Containers.
The motivation comes down to a few key reasons:
- Difficulties with BioContainers
- New features we’d like
- Conda lock files
- Simplified developer workflow
- Better transparency
With the new Seqera Containers setup we should be able to address these problems, as well as provide the additional features.
A detailed description of the proposed mechanism will come in part 2 of this blog post. Here are the key points:
- Developers will only need to edit the conda
environment.yml(no processcontainer) - Container images will be built by Wave and stored in Seqera Containers
- Build logs and source files will be shown on the nf-core website module pages
- Conda lock-files will be used for Conda users and CI tests
- Software releases will be automatically bumped in modules, using Renovate
- There should be almost no change in how end-users run nf-core pipelines
This move has been in the works for over a year now. It started with discussions between nf-core maintainers and Seqera developers about what the community needed. Seqera’s open source Wave container tool brings convenience, but long term storage of container images and stable container URIs were identified as hard requirements. In response, Seqera developed Seqera Containers, a free community resource with hosting infrastructure funded by AWS. This service was launched in spring 2024.
The adoption of Seqera Containers in nf-core was initially raised in the nf-core steering group, followed by discussion in the core team and then maintainers team (see the nf-core governance structure). At each step, the discussion informed the planned infrastructure and setup for this change, as well as the development of Seqera Containers and Wave.
Several steps are still needed to complete this migration:
- nf-core/modules automation for fetching images and pinning conda-lock files and
meta.ymlreferences - nf-core/tools tooling for pipeline config file generation, with pipeline template update
- Bulk update of nf-core/modules to use Seqera Containers
- Update of nf-core pipelines to use the new modules
If all goes to plan, we hope to have all modules updated by the end of 2024. This work will be tracked in GitHub issues as usual, starting with nf-core/modules#5832. This issue has already had a substantial amount of discussion, much of which has informed this blog post.
It’s not too late to add your feedback! So if you have any questions or ideas, let us know on Github or Slack.
Note that images from Seqera Containers can already be used in nf-core/modules, with the old syntax of using a container declaration (see example).
BioContainers and Seqera Containers
Nearly all nf-core modules are bundled with a conda environment.yml, listing software package dependencies from Bioconda (eg. FastQC).
The BioContainers project conveniently builds Docker and Singularity images for all tools on Bioconda automatically, hosting the images publicly on quay.io and the Galaxy FTP servers, respectively.
This means that the nf-core module developer can find the matching BioContainer images for their Bioconda package, and then add the image URLs into the module’s main.nf script (see FastQC example).
The nf-core pipeline user then pulls the container images from quay.io or the Galaxy FTP server when they run the pipeline.
Seqera Containers was launched in spring 2024 (see Nextflow Summit talk, Seqera blog post, AWS blog post + podcast, and the Nextflow Channels podcast for more information).
Seqera Containers allows anyone to request a container image based on Conda or PyPI packages. The image is built on demand and then saved so that subsequent requests return the exact same container image files.
The Seqera Containers service has a lot in common with BioContainers. Both generate Docker and Singularity images from Bioconda. The main difference is when those builds happen.
Seqera Containers is built on top of Wave - an open-source tool developed by Seqera for on-demand generation of containers. When a new tool or version is requested, Wave builds a container using Conda and returns it. In contrast, BioContainers runs a build when a new package is created on Bioconda. Building on-demand gives greater flexibility and scalability, especially for multi-tool containers.
nf-core pipeline users won’t need to interact with Wave directly. We simply plan to replace the mechanism for supplying the default container images specified in pipelines.
It’s important to distinguish the differences between Wave and Seqera Containers. Here’s a brief comparison of the features:
| BioContainers | Wave | Seqera Containers | |
|---|---|---|---|
| Support Bioconda packages | ✅ | ✅ | ✅ |
| Support all conda channels | ❌ | ✅ | ✅ |
| Support PyPI (pip) packages | ❌ | ✅ | ✅ |
| Docker + Singularity support | ✅ | ✅ | ✅ |
| Linux aarch64 and arm64 | ⏳ In progress | ✅ | ✅ |
| Multi-package containers | ✅ Mulled | ✅ | ✅ |
| Conda lock files generated | ❌ | ✅ | ✅ |
| Container build logs | ❌ CI logs short lived | ✅ | ✅ |
| Docker container security scans | ✅ quay.io | ✅ Trivy | ✅ Trivy |
| SBOM manifests (software bill of materials) | ❌ | ✅ | ✅ |
| Long storage duration | ✅ * | ❌ 72 hours cache | ✅ * |
| Pull delay for conda packages | ✅ instant | ❌ ~2-3 minutes for build on first request | ✅ instant |
| Stable image URIs | ✅ | ❌ Single-run | ✅ |
| Software required for end user | Docker / Singularity | Wave CLI / Nextflow, Docker / Singularity | Docker / Singularity |
| Offline support with downloaded images | ✅ | ❌ | ✅ |
| Guaranteed identical conda builds in future image pulls | ✅ | ❌ | ✅ |
* Forever is a long time. Seqera is committing to keeping images for a minimum of 5 years from the time of their creation. BioContainers has no public policy.
Mulled (multi-package) images

BioContainers is primarily set up to have a 1:1 relationship with Bioconda. This is great for bioinformatics tools as an image is created every time a package is published. This works well until you need to use more than one tool in a single process, particularly when the tools are from elsewhere in the conda ecosystem. For example, many bioinformatics tools leverage samtools to convert file formats or sort reads on the fly. Others may pipe output to compression tools like pigz. To resolve this, BioContainers has the concept of “mulled” images (as in mulled wine). Unfortunately, generating mulled containers is not trivial.
Luke Pembleton, Nextflow Ambassador, has a great blog post summarising the dark art of Finding the right mulled biocontainer. Galaxy also has documentation on the topic.
In short, to request an image, you need to scroll to the bottom of a large CSV file full of hashes and add a line with your requested conda packages. Once edited, the images are built on CI. The container then becomes available after a review and merge.
It used to be that you had to hunt through the GitHub action log to find the URI for your new container. However, Moriz E. Beber from the nf-core community has created a webpage to generate the name of the mulled container which gently guides you through the process of finding the name of your container and how to update a container if you want to bump the software versions.
Once the image is built and you know its address, the final step is to go to nf-core/modules,
create a pull-request with the updated containers, and bump the versions in the conda environment.yml.
This system works and, despite its complexity, is now a familiar process to many nf-core developers. However, it does present some problems. It’s a highly manual process to update software versions and the container declarations - just fetching the software for a module often ends up taking longer than writing the entire module. There are also significant delays in waiting for the images to become available. All in all, it can be a frustrating experience and we see this in the volume of Slack messages asking for help. It seems likely that this puts some people off from contributing to nf-core.
Migrating nf-core to use Seqera Containers will make provisioning multi-package containers easier.
The module developer will only need to edit the module environment.yml file and everything else will be fully automated and made available almost immediately.
Packages can be used minutes after their release on Bioconda and we will have automation in place to bump the Conda + Seqera Containers packages when a tool is updated.
Time to Singularity image
BioContainers builds both Docker and Singularity images. Docker images are pushed to quay.io and are available almost immediately, whereas Singularity images are pushed to the Galaxy FTP server in a nightly build, taking up to 24 hours to become available.
This delay is frustrating for developers, as it breaks the flow of working on an update. Once a new version of a tool is released, they have to wait for the Singularity image to become available before they can add it and test the module. In practice, this often means that module updates get stuck in limbo for days until the developer has time to come back and check if the image is available.
Singularity images are generated on the fly with Seqera Containers. Developers will be able update the nf-core module minutes after the Bioconda package is released.
BioContainers API
To simplify adding single-package BioContainer images to nf-core modules, the nf-core/tools package queries the BioContainer API for a given tool version and fetches the image URIs. This process is repeated during linting of nf-core modules and pipelines, to ensure that there is not an accidental mismatch between Conda and Docker / Singularity software versions. Unfortunately, the BioContainer API has had a history of being slow and unreliable, with a lot of down time. This has the knock-on effect of causing many CI tests to fail, which is frustrating and delays the process of pull-request merges.
By migrating to Seqera Containers we will have similar automation when linting modules, but it will use the Seqera Wave API instead. This has been built to scale to very high volumes and has an extensive and robust back-end with a high degree of monitoring. Note that the Wave API will not be used when people run nf-core pipelines, this is only developers running linting and GitHub Actions automation during development.
Reliability of hosting
BioContainer Docker images are hosted publicly on quay.io. This service is provided free of charge, however in recent years we have had some reliability issues. This becomes more noticeable as the community grows and the number of users trying to pull images increases. Because quay.io is a huge service for whom we are only a tiny player, we have no recourse when this happens and have to just wait until it becomes available again.
Seqera serves nf-core as its primary community: they will respond immediately to any problems or needs. All Seqera Container images (Docker and Singularity) are hosted on custom infrastructure built by Seqera with hosting provided by AWS, so the whole stack is within reach.
Key features

Exceptionally reproducible
Seqera container images are hosted on long-term infrastructure and because their URLs will be hardcoded into pipeline configuration, they should be highly reproducible. Even if new builds of the same software are created in the future using Wave (for example due to an update in the base infrastructure used by Wave) then the old images are still pinned by the pipeline and will continue to be used, much like the BioContainer URIs used currently.
As container URIs will be stored in the module-level meta.yml file, any pipelines using the same shared nf-core/module will also be using the exact same container images.
We will further improve reproducibility at the conda level by adopting the use of conda lock-files. These pin the exact dependency stack used by the build, not just the top-level primary tool being requested. This effectively removes the need for conda to solve the build and also ships md5 hashes for every package. This will greatly improve the reproducibility of the software environments for conda users and the reliability of Conda CI tests.
# This file may be used to create an environment using:
# $ conda create --name <env> --file <this file>
# platform: linux-64
@EXPLICIT
https://conda.anaconda.org/conda-forge/linux-64/_libgcc_mutex-0.1-conda_forge.tar.bz2#d7c89558ba9fa0495403155b64376d81
https://conda.anaconda.org/conda-forge/linux-64/libgomp-14.1.0-h77fa898_1.conda#23c255b008c4f2ae008f81edcabaca89
https://conda.anaconda.org/conda-forge/linux-64/_openmp_mutex-4.5-2_gnu.tar.bz2#73aaf86a425cc6e73fcf236a5a46396d
https://conda.anaconda.org/conda-forge/linux-64/libgcc-14.1.0-h77fa898_1.conda#002ef4463dd1e2b44a94a4ace468f5d2
# .. truncated ..Trust and transparency
Reproducibility is one thing, but it’s also important that the images we use can be trusted and are secure.
Users and developers must be able to verify that the contents of the container match the environment.yml for the module.
The first point of trust is the community.wave.seqera.io base URI.
This is only used for Seqera Containers and only wave.seqera.io is able to push images.
It is also more locked-down than regular Wave, only allowing Conda builds and not custom Dockerfiles or augmented images.
Second, the image URI includes a tag that has a hash of the input files used to request the image. We can use this to retrieve build details, which we will add to the nf-core website module page:
- Build time, architecture and platform (Docker / Singularity)
Dockerfile/ Singularity recipe and Condaenvironment.yml- Full build logs
- Conda lock files with full package URLs and md5sums for entire dependency resolution
- For Docker images: Trivy security scan and software bill of materials (SBOM)
We haven’t built this yet, but here’s what the Wave build details page looks like, which has the same information:
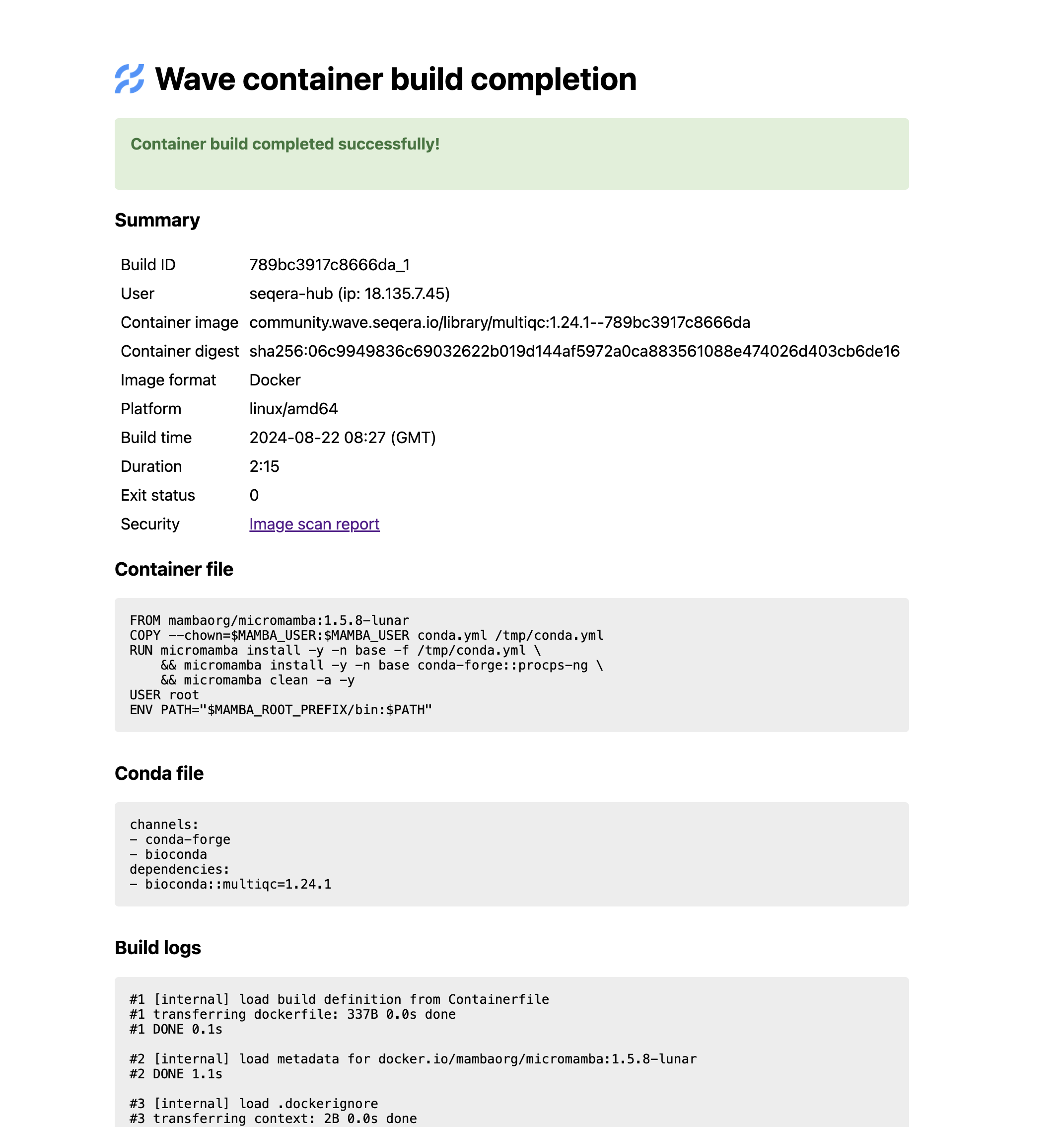
Wave build details page for the MultiQC v1.24.1 image: community.wave.seqera.io/library/multiqc:1.24.1--789bc3917c8666da
The final part is the hash - from this we can infer the Wave Build ID and retrieve the build details: https://wave.seqera.io/view/builds/789bc3917c8666da_1
The new conda lock files also allow enhanced reproducibility for Conda users and more stable CI tests - we’ll come back to these in a future blog post / bytesize.
Finally, the image creation will happen automatically on GitHub Actions when an environment.yml file is edited.
This means that the entire flow from conda file through to final containers will be entirely transparent, as the request to Wave itself and its response will be available in the GitHub Actions logs.
No lock-in
Using Seqera Containers does not mean that nf-core will be locked into using Seqera tooling. The suggested implementation has two components:
- Automated on-demand generation of container images using Wave
- Long term hosting images on Seqera Containers
The tooling for image generation is open-source. Seqera is hosting this API and build service for the community for free, but there’s nothing to stop us from hosting the same API ourselves in the future. As such, there is no lock-in on this API or any tooling we will write around it.
Long-term hosting is more fixed, but again Wave is designed to work with any container registry. So if we want to change in the future we simply flip a configuration value and the generated images will be stored (“frozen”) at an alternative registry of our choosing. Old pipelines would have their configuration overwritten to use a different base registry.
This is the same situation that we have currently with BioContainers and quay.io / Galaxy hosting. The nf-core community will remain free to pick and choose hosting solutions as needed.
Special cases
Not all tools can use Biocontainers, and we’re aware of some special-case packages that have only bespoke Docker images. These will continue to work as they do today.
For people who need to mirror container images to their own custom registry, this will still be possible. Changing the registry base will continue to work exactly the same way it does today.
What this means for you
Users
If you’re using nf-core pipelines but not developing them, then nothing should really change! All nf-core modules and pipelines should start supporting linux/arm64 CPU architectures, such as AWS Graviton / Raspberry Pis(!). Conda users should benefit from faster environment resolution, with more reproducible and stable software thanks to use of the lock files. Finally, the failure rate of docker pulls will improve as we phase out our usage of quay.io.
Developers
If you’re a maintainer of a pipeline that uses nf-core/modules, this means that your life is about to get easier! Never again will you need to try to figure out how to make a mulled image, or wonder when your Singularity image build will be available. New software releases can be used with modules within minutes of release and most updates should happen automatically - freeing up maintainers from having to deal with routine package updates and reviews.
In part-two of this blog post we will dive into the details of how we intend to build this tooling infrastructure in nf-core, so please check that out if you’re interested.
In conclusion
We hope that the nf-core community is excited about these proposals, if you have any questions or concerns then please let us know in the nf-core Slack ❤️
